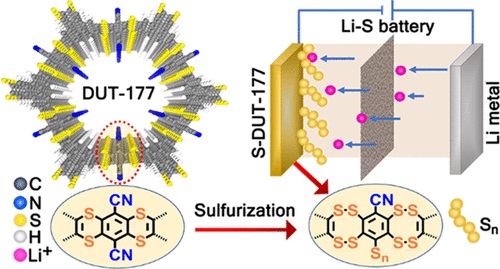
该论文在通过动态自修复的亲核芳族取代反应形成二硫胺键,自下而上合成多孔噻蒽基二维共价有机骨架 (COF)。该论文首次将这种有机硫部分作为结构构件整合到结晶层状 COF 中。新材料的结构与COF典型的平面层间π-堆叠不同,形成了由沿C-S-C键弯曲引起波纹层,但整个COF结构的芳香性和结晶度没有损失。对 COF 和具有噻蒽部分的模型化合物的综合实验和理论研究表明,硫孤对电子在 COF 芳香骨架上的部分离域降低了带隙并提高了氧化还原活性。合成后硫化过程允许多硫化物直接共价连接到骨架的碳主链上,从而为具有最小化多硫化物穿梭的锂硫 (Li-S) 电池提供分子设计的正极材料。即使在 500 mA/g 电流密度下进行 500 次充放电循环后,电池仍能提供近 77% 的初始容量。这种新型硫键是COF化学中理想的模型材料,是在分子水平上研究锂硫电池的先进电极材料。
结晶层状共价有机骨架(COFs)是由高度对称的芳香族单元组成的多孔材料,这些芳香族单元通过共价键在二维(2Ds)内连接,并通过π-π相互作用堆叠成三维(3D)。从COF的发展历史上看,从环硼氧烷和硼酸酯开始,扩展到多种不同的有机链接键,包括亚胺和 sp2C-sp2C 连接单元,( 2D ) 框架的构建块在很大程度上取决于这些不同链接中的轨道对称性。因此,整体 COF 骨架及其结构相关性质受其连接键的支配。例如,从 C=C 和 C=N 键切换到基于稠合芳环的键可增强 COF 层的结构刚性并有助于整个结构的电子共轭。此外,由芳香杂环组成的键构建二维结晶 COF 使所得材料具有氧化还原特性。这种芳族杂环的突出例子包括二恶英、吩嗪、恶唑、噻唑、苯并咪唑、或苯并恶唑键,这会影响所得材料的能带结构的能级。这为通过网状化学精确地设计框架带隙提供了机会,以便在诸如光催化、电催化、电子设备和能量存储等应用中使用类似策略。
在含硫杂环的情况下,空缺的硫 d 轨道可能与客体离子相互作用来存储电荷,硫的低电负性也为杂环的 C-S 键提供孤对电子。这对于电子共轭发挥重要作用,尤其是当它以自由基阳离子形式存在时。此外,适度的 C-S 键强度通过合成后转化促进化学可调性。然而,富硫骨架的稳定性差以及含硫前驱体的不稳定性阻碍了杂环闭环。虽然含恶唑、噻唑和苯并恶唑的二维 COF 遵循类似的闭环,但它们主要由邻羟基/硫醇取代的芳基胺构成。而沿单键的自由旋转会破坏层中的电子共轭,与此相反,通过二酮与邻二胺反应形成的吩嗪键产生芳族和平面骨架,但由于环化过程中的低可逆性导致的相对低的结晶度,长程周期有序受到阻碍。
由于连接的稠合芳环使框架能够承受合成后修饰的苛刻条件,因此通过环化形成的 COF 提供了额外的稳定性。此外,含有 p 个嵌段元素(O、S、Se 或 P)的稠合杂环将推动 COF 向利用其电子特性的应用发展。设计合适的联结和优化 COF 构建的反应条件对于合成化学家来说仍然是一项艰巨的任务。Yaghi 及其同事在 COF-316 的合成中建立了通过二醇和二氟官能化连接分子之间的不可逆亲核芳族取代 (SNAr) 在二维 COF 中形成二恶英键。这是稠合杂环有机键建立结晶且稳定的 COF 的例子,它已被用于从催化到能量存储设备的各种应用。转换这种不可逆的闭环机制以开发具有其他杂环键的结晶 COF 仍然是一个的挑战。
合成与表征
在本文中,作者合成了基于噻蒽结构的 COF:DUT-177 。其结构模型在计算机上进行了模拟,并通过粉末 X 射线衍射 (PXRD) 进行了实验验证(图 1C 和 2),证实了类似于已知的二恶英连接的 COF-316 的周期性蜂窝结构的形成。Brunauer-Emmett-Teller (BET) 表面积为 709 m2 g-1。
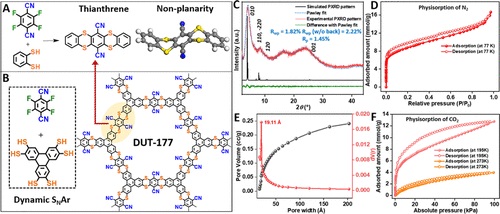
图 1:DUT-177合成示意图以及结构表征
这两个 S-C 键的面外弯曲具有 101° 的二面角,理论计算研究的 DUT-177 分子单元验证了面外弯曲,根据噻蒽排列(噻蒽环的上下弯曲和全上下弯曲)。这两种异构体可以通过克服 5.06 kcal mol-1的低能垒可逆地互换。虽然层的对称叠加在C2/m空间群中建立了周期结构,但密度泛函理论结果表明,AA倾斜叠加几何结构在P1空间群中是能量有利的。并且其计算的 PXRD 峰形与实验 PXRD 非常吻合。有趣的是,在这种最低能量结构中,该层围绕噻蒽单元中的碳硫键弯曲,并在两个不同方向上弯曲,形成波浪状的二维堆叠(图 2B)。来自相邻层的噻蒽和苯并苯单元在 3.93 Å 的距离处 π 堆叠(不包括原子的范德华半径),在整个结构中保持相同的层距离并产生一维纳米通道。然而,2D 结构的 3D 传播源自固定在 DUT-177 孔壁上的具有大量极性官能团(含硫和含腈的环)的众多微孔。
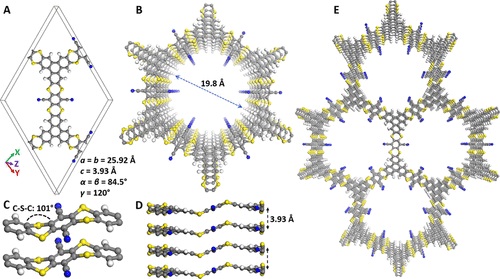
图 2:DUT-177空间结构
二硫胺键影响 COF 的电子能级并在其硫中心触发氧化还原活性。COF的氧化还原活性和电子转移机制主要取决于结构单元轨道的电子密度分布。仔细观察 DUT-177 分子单元的轨道能量图表明,最高占据分子轨道 (HOMO) 在 π 共轭骨架上离域,而最低未占据分子轨道 (LUMO) 是位于腈取代的苯环上。通过HSE06 功能得到了(图 3B) 2.45 eV 的实际带隙,这与使用循环伏安法 (CV) 测量得到的 2.33 eV 的实验值非常吻合。考虑到 COF 的 UV-vis 吸收的最大波长的 Tauc 图显示了带边缘的位置为 2.16 eV,这也与理论带隙相当。另一方面,电子密度沿硫桥的强烈离域使芳环电流在整个 DUT-177 的 2D 层中相互连接。与 300 和 800 MHz 的模型化合物相比,这导致 COF 的 13C NMR 信号的谱线显着展宽和分辨率较差。使用自然键轨道 (NBO)分析噻蒽分子的轨道对称性表明,其中一对孤对硫与 s (65.38%) 和 p (34.61%) 杂化,而另一对保留其纯 p特点,噻蒽分子的这一特征与噻吩的特征相似,具有强烈离域的 p 轨道。值得注意的是,噻蒽核心还表现出硫的非键电子之一与相邻碳原子的 2p 轨道的重叠,但程度较弱。
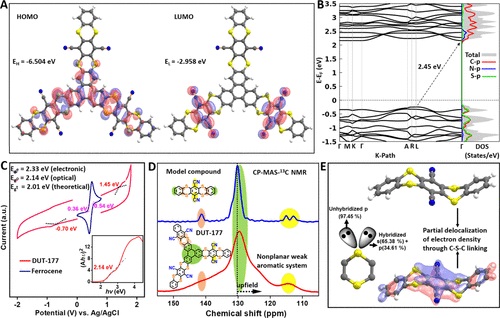
图 3:(A) DUT-177分子单元上HOMO和LUMO的分布;(B) 使用HSE06函数计算AA倾斜堆叠DUT-177的电子能带结构分析;(C) DUT-177非水CV测量的氧化还原电位表示HOMO和LUMO的能量以及电化学能隙。插图:从DUT-177的紫外-可见吸收光谱获得的Tauc图,用于确定光学带隙;(D) DUT-177及其模型化合物芳香区的CP-MAS-13C NMR;(E) 从理论上计算了硫在二硫键中的可用孤对杂交。
噻蒽结构的硫化
硫中心的氧化还原亲和力和框架结构上的高电子密度使 DUT-177 成为电荷存储的理想候选者。然而,由 DUT-177 和锂金属参比电极制成的典型锂离子电池半电池在 1.75 V 时表现出不可逆的 COF 降解,这可能归因于电极上的碳-硫键断裂和不可逆的锂盐沉积。另一方面,二硫键属于有机聚硫烷类,其中一个硫原子在两个碳原子之间共价共享。在这样的系统中,硫-硫亲和力对于在硫化反应过程中将有机硫化合物相互转化为更多含硫原子的类似物至关重要。为了研究二硫胺键转化的可能性,模型化合物在 180 °C 和真空条件下用过量硫处理。在众多可能性中,转化发生在硫桥本身以生成二噻蒽结构,硫化后噻蒽可能转化为二噻蒽,这是对这种转化的首次报道。所得材料失去其结晶性,但通过 PXRD 分析也未观察到结晶元素硫杂质。X 射线光电子能谱 (XPS) 和红外光谱证实了通过部分取代腈基团的多硫化物共价锚定,而俄歇电子能谱 (AES) 和拉曼研究检测了DUT-177 和 S-DUT-177从噻蒽到二噻蒽结构的转化。
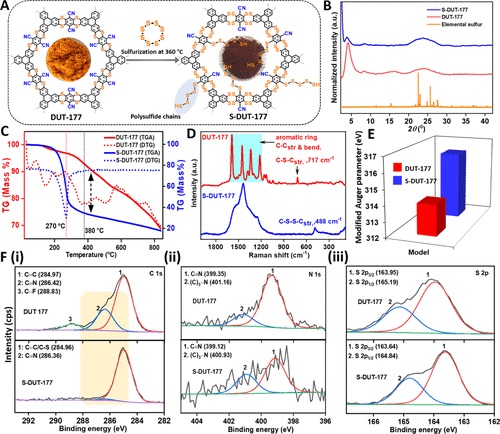
图 4:(A) DUT-177 硫化的示意图;(B) PXRD;(C) TGA-DTA 图;(D)拉曼位移;(E) 从俄歇电子能谱(F) X 射线光电子能谱分析数据
锂硫电池中共价锚定多硫化物的活性
这些硫连接的COF(噻吩和二噻吩结构)中存在噻吩和腈,表明在较高的电位窗口(1.2至2.8 V)中对锂金属电极具有阴极活性。从DUT-177电池和锂金属参比电极的循环伏安图显示,在所有分析的扫描速率下,在1.75 V下,当在二氧杂环乙烷(DOL)/二甲氧基乙烷(DME)(1:1)中使用锂双-(三氟甲基磺酰基)酰亚胺(LiTFSI)和0.1 M硝酸锂(LiNO3)作为电解液时,会发生显著而强烈的氧化。S-DUT-177 的二噻蒽结构和锚定的多硫化物与锂离子与来自二硫键的可用孤对电子可逆地相互作用(“S-S”键)。
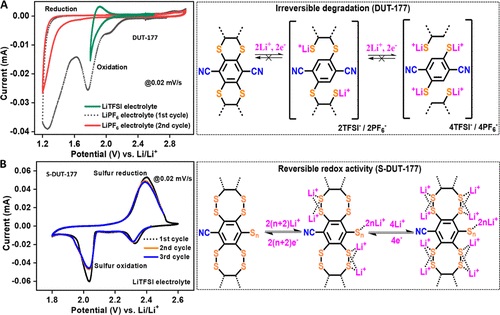
图 5:(A) DUT-177电池的 CV (B) S-DUT-177电池的 CV 测量显示可逆的氧化还原;(右)S-DUT-177 中锂离子与共价锚定硫的可逆相互作用机制的图示
锂离子与多个硫原子的相互作用证实了将这种硫化COF(S-DUT-177)用于先进锂硫电池的潜力。阻抗分析的奈奎斯特曲线图证实,S-DUT-177衍生币形电池在极低频区域(0.1 MHz)的交流频率较高,但锂的扩散率较差,其电阻率较低(90Ω)。锚定的多硫化物链的氧化还原活性主要有助于充电 - 放电曲线,具有 2.08 V 的长程电压平台。随着电流密度的增加,从2.08 V电压平台获得的比容量逐渐降低(在1 a/g时,比容量为276 mA h/g)(图6C,D)。有趣的是,与大多数硫浸渍多孔COF相比,聚硫锚定S-DUT-177的速率性能有所改善。在 500 次循环后,纽扣电池在以 500 mA/g 运行时(图 6E),仍能提供 76.6% 的初始容量(图 6E),这大大优于“COF–F–S”(100 次循环后保留 64.5%)。因此,S-DUT-177 中的共价锚定硫实体可以抑制初始循环中的穿梭效应(Li-S 电池中的多硫化物溶解)。
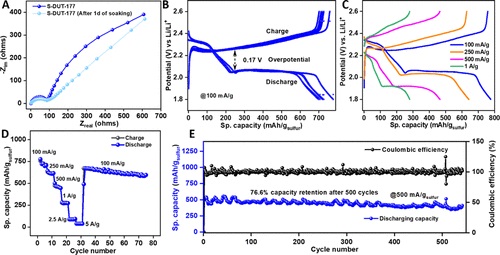
图 6:(A)奈奎斯特图;(B,C)S-DUT-177电池在不同电流密度下的充电-放电曲线;(D) S-DUT-177电池在不同电流密度下的速率性能;(E) S-DUT-177电池在500 mA/g电流密度下的循环稳定性
DUT-177闭环反应导致二硫键连接的框架具有微弱的芳香环电流以及COF层之间的波浪型堆叠相互作用,并具有独特的电子特性。这不仅带来了新的连接方式,扩展了2D层状COF家族,还为合成后修饰提供了一个动态平台。噻蒽COF的硫化显示了二噻蒽结构的相互转化以及通过腈部分取代共价锚定多硫化物的可能性。由于锂与硫化骨架的稳定可逆相互作用,这使得硫实体具有氧化还原活性。然而,硫化降低了骨架的结晶度,阻碍了可能的锂骨架-硫相互作用的机理研究。改进设计策略和开发合适的连接方式可以提高框架中固有的硫含量,以满足改善锂硫电池阴极活性的要求。通过多硫化物环将硫与骨架紧密结合,或使用多硫化物本身作为骨架结构的连接,可以潜在地减少较高电化学电位窗口中的穿梭效应。含有硫键的高级COF在分子水平上对锂硫电池的理解更加深入。特别是电子导电框架将显著改善电池的整体性能。
Porous Dithiine-Linked Covalent Organic Framework as a Dynamic Platform for Covalent Polysulfide Anchoring in Lithium–Sulfur Battery Cathodes
Sattwick Haldar, Mingchao Wang, Preeti Bhauriyal, Arpan Hazra, Arafat H. Khan, Volodymyr Bon, Mark A. Isaacs, Ankita De, Leonid Shupletsov, Tom Boenke, Julia Grothe, Thomas Heine, Eike Brunner, Xinliang Feng, Renhao Dong, Andreas Schneemann, and Stefan Kaskel
DOI:10.1021/jacs.2c02346
原文链接:https://pubs.acs.org/doi/10.1021/jacs.2c02346


 购销咨询
购销咨询 
